
Mutation score
Contents
Mutation score¶
To compute the mutation score, for a given mutation we can consider the amino acid in the wildtype protein as a reference state, and compare the probability assigned to the mutated amino acid with the probability assigned to the wildtype.
In practice, at each mutated position, we introduce a mask token and record the model’s predicted probabilities of the tokens at that position. This metric is describe in the paper
We call it the masked marginal probability, which is described below:
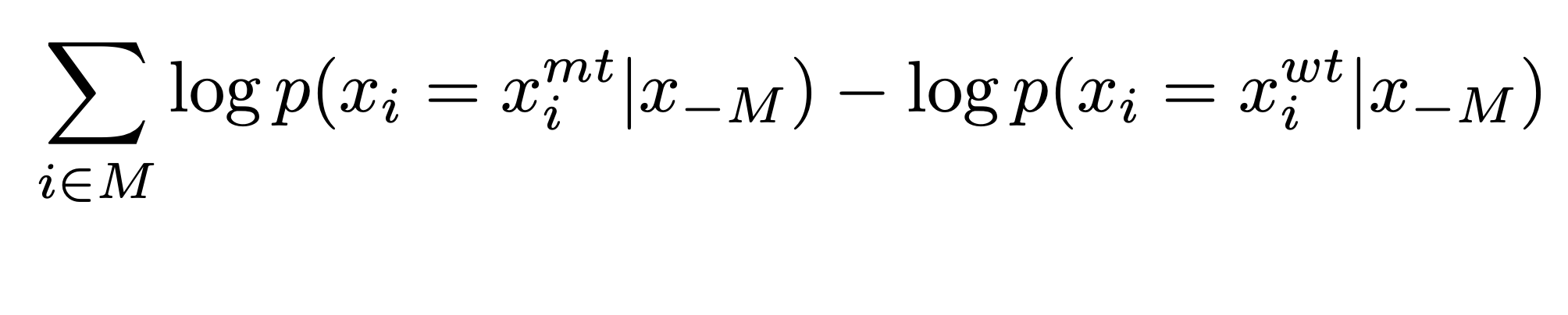
compute_mutation_score function¶
To compute this metric, we need to provide to things:
sequences: a list of sequence to score
mutations: a list of sequence’ mutations. A sequence” mutations can be composed of multiple single mutations.
Example¶
from biotransformers import BioTransformers
sequences = ["MAPSRKFFVGGNWKMNVVCAPPTAYIDFARQKLDPKI",
"AVAAQNCYKVTNGAFTGEISPGMIKDCGATWVVLGH",
"GRKQSLGELIGTLNAAKVPADTE"]
mutations = [["M1P","K6F","N12G"],["A4V"],["A15L","V18A"]]
bio_trans.compute_mutation_score(sequences,mutations)
>> [-4.07569046318531, -0.848480224609375, 0.4472615122795105]
To create a mutations list, you need to respect the mutations format above in the example:
format : Native_aaPositionMutated_aa.
Important
Your position index for mutation must start at 1.